Antibacterials are antimicrobial agents particularly used to kill bacteria. These includes naturally occurring as well as synthetic and semisynthetic compounds.
Antibiotics are chemicals produced by microorganisms that, at extremely low concentrations, kill or selectively limit the growth of other microorganisms without having a substantial negative impact on the host. Other naturally occurring compounds that inhibit microorganisms but are created by higher forms (such as antibodies) or even by microbes but require high quantities (such as ethanol, lactic acid, H2O2) are not included by this definition.
Chemotherapeutic agent: This term was formerly restricted to synthetic compounds, but since numerous antibiotics and their analogues have been synthesised, this criterion has become irrelevant. It would be more meaningful to use the term antimicrobial agent (AMA) to designate synthetic as well as naturally obtained drugs that attenuate microorganisms.
Natural sources include bacteria (bacitracin, colistin, polymyxin B, aztreonam), fungi (penicillin, cephalosporin, griseofulvin) and actinomycetes (tetracyclines, chloramphenicol, aminoglycosides, macrolides).

History
With the discovery of microbes in the late 19th century and that they are the cause of many diseases, Ehrlich experimented with the assumption that if particular dyes could selectively stain microorganisms, they could also be selectively toxic to these organisms. He tried methylene blue, trypan red, etc. He developed the arsenicals—atoxyl for sleeping sickness, arsphenamine for syphilis. He coined the term ‘chemotherapy’ because he used drugs of known chemical structure (that of most other drugs in use at that time was not known).
The modern era of chemotherapy was pioneered by Domagk in 1935 by demonstrating the therapeutic effect of Prontosil (a sulfonamide dye) in pyogenic infection. It was soon realized that the active moiety was para-amino benzene sulfonamide, and the dye part was not essential. Sulfapyridine was the first sulfonamide to be marketed in 1938.
Pasteur first showed the concept of antibiosis in 1877, showing how airborne bacteria could inhibit the growth of anthrax bacilli in urine. Fleming (1929) discovered that Penicillium mould produced a diffusible material that could kill Staphylococcus on the culture plate. He named this substance ‘penicillin’ but could not purify it. Chain and Florey followed up this observation in 1939 which culminated in the clinical use of penicillin in 1941.
In the 1940s, Waksman and his colleagues began an organised search of Actinomycetes as source of antibiotics and discovered streptomycin in 1944. This group of soil microbes proved to be a treasure-house of antibiotics and soon tetracyclines, chloramphenicol, erythromycin and many others followed.
In the past 50 years emphasis has shifted from searching new antibiotic producing organisms to developing semisynthetic derivatives of older antibiotics with more desirable properties or differing spectrum of activity. Few novel synthetic AMAs, e.g., fluoroquinolones, oxazolidinones have also been produced.
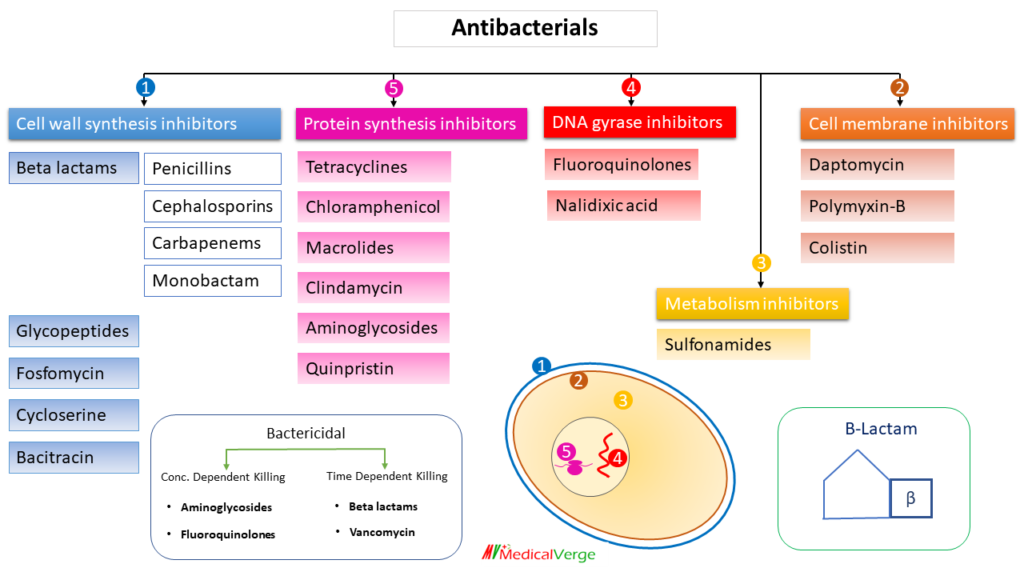
Antibacterial Classification
Antibacterial agents can be classified in many ways. e.g., on the basis of –
1) organism (anti-tubercular, anti-leprotic, anti-pseudomonal etc.)
2) spectrum (against gram positive/negative),
2) source (antibiotics and non-antibiotics),
3) chemical structure (aminoglycosides, quinolones, macrolides etc.)
4) type of action (cidal and static),
5) mechanism of action etc. herewith classification on the basis of mechanism –
Cell wall synthesis inhibitors
1. Beta-lactams
All beta lactams sharing a common structure of beta lactam ring, so named as. b-Lactam antibiotics produce bactericidal effect by inhibiting cell wall synthesis. Bacterial cell wall is composed of peptidoglycan which contains amino sugars, N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG). The enzyme, transpeptidase (a PBP), removes terminal alanine of one strand resulting in its linkage with glycine of adjacent strand. Cross-linking makes the cell wall rigid and stable. beta-Lactams, the structural analogues of d-alanine, inhibit transpeptidase, thus inhibiting cross-linking of peptidoglycans and cell wall synthesis. The cell wall in gram-positive bacteria is composed mainly of highly cross-linked peptidoglycan, which is 50–100 layers thick and is near the cell surface. In gram-negative bacteria, the peptidoglycan layer is only one to two molecules thick. Hence, gram-positive organisms are more susceptible to penicillin.
a) Penicillins
Penicillin was accidentally discovered by Alexander Fleming. It was originally obtained from the fungus penicillium notatum, but the present source is a high yielding mutant of penicillium chrysogenum. All penicillin structure consists of a sulfur containing thiazolidine ring, fused with a beta-lactam ring to which a side chain is attached at position-6. A variety of semisynthetic penicillins are synthesized by attaching appropriate side chain at position-6.
Penicillin-G (PnG)
The first penicillin to be obtained naturally was Penicillin-G (benzyl-penicillin). Salts of Na+ and K+ are more stable than parent acid. Sodium PnG is highly water soluble. It is stable in the dry state, but solution deteriorates rapidly at room temperature, though it remains stable at 4°C for 3 days. Therefore, PnG solutions are always prepared freshly. 1 U of crystalline sod. benzyl penicillin = 0.6 μg of the standard preparation. Accordingly, 1 g = 1.6 million units. PnG preferably given by IV route and less commonly by IM route because of pain at injection site.
There are various limitations of PnG –
- Orally not effective, because PnG is acid labile.
- Short acting: 4-6 hours (excreted by active tubular secretion, so required to administer several times in a day)
- Narrow spectrum (effective against gram-positive only)
- Resistance development
- Allergy in many people
To overcome most of the above drawbacks, semisynthetic penicillins have been developed –
Oral penicillins (acid stable)
- Penicillin-V (phenoxy-methyl penicillin)
- Oxacillin
- Cloxacillin
- Dicloxacillin
- Ampicillin
- Amoxycillin
Long-acting preparations
- Penicillin excreted by tubular secretion. Probenecid inhibits tubular secretion, thus on combining with it penicillin become long acting.
- Depot preparation: benzathine penicillin (3-4 weeks) and procaine penicillin (12-24 hours), hold it in muscle (given I.M.) to make it longer acting by releasing slowly. If this IM depot preparation is given IV, they become short acting.
Extended spectrum penicillins
- Ampicillin
- Amoxycillin
- Carbenicillin
- Ticarcillin
- Azlocillin
- Mezlocillin
- Piperacillin
Combining with beta-lactamase inhibitors to overcome resistance by beta-lactamase enzyme.
- Clavulanic acid (with amoxycillin)
- Sulbactam (with ampicillin)
- Tazobactam (with piperacillin)
*Avibactam (with ceftazidime)
Beta-lactamase (penicillinase) resistant penicillins
- Oxacillin
- Cloxacillin
- Dicloxacillin
- Nafcillin
- Methicillin
*Staphylococcus aureus may develop resistance to methicillin, by altered penicillin binding protein (PBP). This bacterial strain named as MRSA, which is resistance to all beta-lactam antimicrobials except 5th generation cephalosporins.
To avoid allergy
Penicillins can cause immediate type of hypersensitivity reaction (type I hypersensitivity). It is not a dose-related adverse drug reaction and can occur with any dosage form of penicillin. Cross-reactivity can occur among penicillins and also among other b-lactam antibiotics except monobactam.
Before giving penicillin, hypersensitivity testing is done by intradermal route on ventral aspect of forearm.
b) Cephalosporins
The first cephalosporins were obtained from a fungus, Cephalosporium acremonium. Later, semisynthetic cephalosporins were developed. Cephalosporins are b-lactam antibiotics with 7-aminocephalosporanic acid nucleus. The mechanism of action and development of resistance are similar to those of penicillins. Like penicillins, cephalosporins also inhibit synthesis of bacterial cell wall and produce bactericidal effect. Cephalosporins have been divided into five generations –
1st generation
They are active against gram positive only, similar to penicillins.
- Cefazolin
- Cefalexin
- Cephalothin
- Cefadroxil
- Cefaloridine
2nd generation
Activity against: mainly gram negative, as well as anaerobes.
- Cefaclor
- Cefuroxime
- Cefoxitin
- Cefmetazole
3rd generation
Activity against: mainly gram negative, as well as gram positive.
- Cefixime
- Ceftriaxone
- Cefoperazone
- Cefpodoxime
- Ceftazidime
- Ceftibuten
4th generation
Activity against: mainly gram negative
- Cefepime
- Cefpirome
5th generation
Reserved for MRSA.
- Ceftaroline
- Ceftobiprole
c) Carbapenems
Carbapenems are beta-lactamase resistant bactericidal agent. It has broad spectrum activity: against gram-positive, gram-negative as well as anaerobes. Imipenem is the first antimicrobial agent in this class, which is ineffective in vivo because of broken down by a renal enzyme dehydropeptidase. Hence always given in combination with cilastatin, which is a dehydropeptidase inhibitor. Imipenem can cause seizure as a side effect. Because of these 2 limitations of imipenem, newer carbapenems were developed, which are dehydropeptidase resistant and have lesser risk of causing seizure –
- Meropenem (I.V.)
- Doripenem (I.V.)
- Ertapenem (I.V./I.M.)
- Faropenem (oral)
d) Monobactam
Aztreonam is the only AMA in this class. Only single ring (beta-lactam ring) in its structure, hence named as monobactam. Only effective against gram-negative. It has no cross allergy with other beta-lactam, possibly due to no second ring on its structure.
2. Glycopeptides
Vancomycin is the first agent in this class. It is discovered in 1956 as a penicillin substitute which assumed special significance due to efficacy against MRSA. They are not effective orally, because not absorb from GIT due to large molecular size.
Vancomycin is drug of choice for MRSA infection. On IV administration, it releases histamine leading to red-man syndrome as a side effect. It is given orally for pseudomembranous colitis, where action is locally in GIT.
Other AMAs in this class are following, which have similar gram-positive effectivity but longer acting and do not cause red-man syndrome –
- Teicoplanin
- Oritavancin
- Dalbavancin
- Telavancin
3. Bacitracin
It is one of the earliest discovered antibiotics from a strain of Bacillus subtilis. it is active mainly against gram-positive organisms (both cocci and bacilli). It acts by inhibiting cell wall synthesis at a step earlier than that inhibited by penicillin. It is bactericidal.
Bacitracin is not absorbed orally. It is not given parenterally because of high toxicity, especially to the kidney. Use is restricted to topical application for infected wounds, ulcers, eye infections; generally, in combination with neomycin, polymyxin, etc.
Protein synthesis inhibitors
All protein synthesis inhibitors are bacteriostatic, except aminoglycosides which are bactericidal.
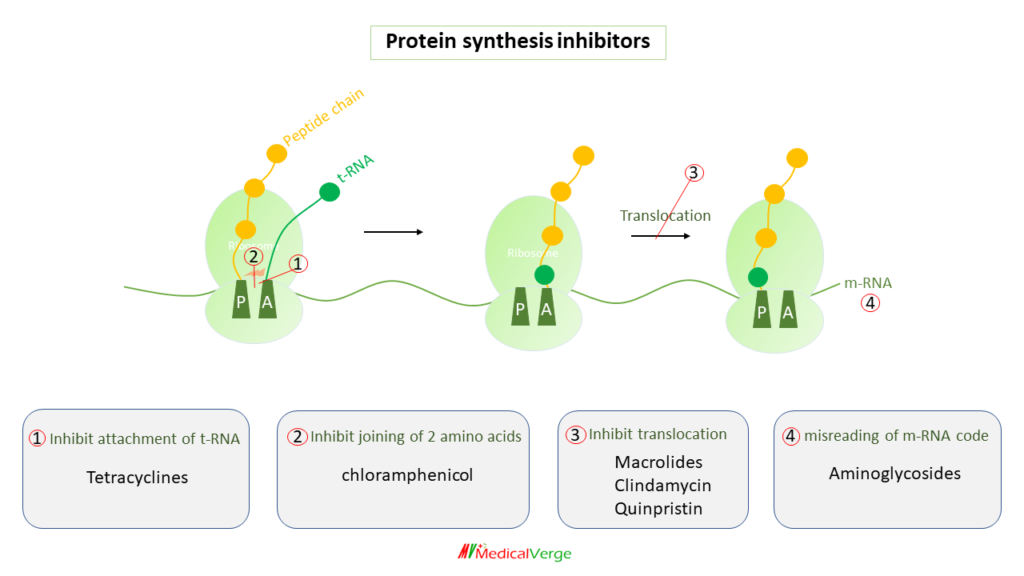
1. Tetracyclines
Tetracycline is so named because its structure consists of four cycles (rings). Tetracyclines are all solids with a mild bitter taste. Tetracycline enters in sensitive bacteria by active transport and in gram negative bacteria diffuse through porin channels as well. Doxycycline and minocycline, which are more lipid-soluble molecules, enter through passive diffusion also (this is partly responsible for their higher potency). It was known as a “broad spectrum antibiotic” because it differed noticeably from penicillin and streptomycin (the other two antibiotics available at the time) in being active orally and in affecting a wide range of microorganisms.
Tetracyclines, which are mainly bacteriostatic, bind to the 30S ribosome and inhibit attachment of t-RNA on acceptor (A) site. It is drug of choice for rickettsial infections (epidemic typhus, Rocky Mountain spotted fever, scrub typhus and Q fever), chlamydial infections (inguinale granuloma, lymphogranuloma venereum) and cholera. Other uses include atypical pneumonia, malaria and leprosy. Doxycycline is preferred for most of the indications of tetracycline. The tetracyclines available in India for clinical use are –
- Tetracycline
- Oxytetracycline
- Demeclocycline
- Doxycycline
- Minocycline
Side effects:
- Phototoxicity (rashes of sun exposure) – highest with demeclocycline
- Diabetes insipidus – highest with demeclocycline
- Deposition on growing bone and teeth (hence contraindicated in pregnancy and children less than 5 years)
- Vestibular dysfunction (highest with minocycline)
- Fanconi syndrome after taking expired drug – In contrast to many drugs, which lose their efficacy after expiration and typically do not produce side effects. Tetracycline has side effects that become apparent after expiration that would not typically exist.
The efflux pump is the main mechanism of bacterial resistance to tetracycline. Tigecycline is a newer drug (glycylcycline), which is resistance to efflux pump, given by I.V. route.
2. Chloramphenicol
Streptomyces venezuelae was used to produce chloramphenicol for the first time in 1947. It was subsequently chemically synthesised, and the current commercial product is entirely synthetic. It inhibits combining of two amino acids (between the newly attached amino acid and the nascent peptide chain), thus it inhibits peptide chain elongation.
It has almost the same range of antibacterial activity as tetracyclines and is a broad-spectrum antibiotic. It was previously the drug of choice for enteric fever (typhoid). Due to resistance that has developed in the majority of cases and its substantial adverse effects, including as bone marrow suppression and grey baby syndrome (cyanosis of newborn), chloramphenicol is rarely used today.
3. Macrolides
These are antibiotics having a macrocyclic lactone ring with attached sugars. Erythromycin is the first member discovered and was isolated from Streptomyces erythreus in 1952. Later on, semisynthetic derivatives were developed. They inhibit translocation of ribosome along the mRNA. Macrolides are 2nd line drugs to penicillin for gram-positive (if allergic to penicillins). Macrolides are DOC for atypical pneumonia, pertussis, chancroid and legionella. Main side effects include diarrhea (as motilin receptor agonist) and reversible ototoxicity.
- Erythromycin
- Clarithromycin
- Roxithromycin
- Azithromycin – longest acting macrolide
- Fidaxomicin – a newer macrolide recently approved for mild to moderate pseudomembranous colitis.
- Spiramycin –a macrolide chemically, used to prevent transmission of Toxoplasma gondii from mother to foetus.
- Tacrolimus – a macrolide chemically, used as immunosuppressant
*Azithromycin has least microsomal enzyme inhibitor property among macrolides.
4. Clindamycin
It is chemically lincosamide, similar mechanism of action and spectrum of activity as erythromycin. However, the distinctive feature is its high activity against a variety of anaerobes. Clindamycin is well absorbed orally. It penetrates into most skeletal and soft tissues, but not in brain and CSF. Topically it is used for infected acne vulgaris. Clindamycin is the most common cause of pseudomembranous colitis. it is secreted in bile, hence safe in renal failure. Additionally, it is effective against parasites such as pneumocystis, plasmodium and toxoplasma.
5. Quinpristin
Quinpristin is always given in combination with dalbopristin, as together exert synergistic inhibition of bacterial protein synthesis. Both are chemically streptogramins and act by inhibiting translocation of ribosome along the mRNA. Antibacterial spectrum is gram-positive cocci including MRSA (Methicillin-resistant Staphylococcus aureus) and some VRSA (Vancomycin-resistant Staphylococcus aureus).
6. Aminoglycosides
These are natural (by soil actinomycetes) and semisynthetic bactericidal agents, contains two or more amino sugars attached with glycosidic bond in their structure. Due to their strong polarity, they are not well absorbed from the GI tract. Parenteral infusion is the route of administration. Unlike penicillin, which was a chance discovery, aminoglycosides are products of deliberate search for drugs effective against gram-negative bacteria. Streptomycin was the first member discovered in 1944. they inhibit protein synthesis by misreading of mRNA code.
Transport of aminoglycoside into the bacterial cell is oxygen dependent active processes; hence, aminoglycosides are not effective against anaerobes. They are effective against gram-negative aerobes including pseudomonas. All exhibit nephrotoxicity (max by neomycin) and ototoxicity (auditory ototoxicity max by amikacin and vestibular ototoxicity max with streptomycin. They also have neuromuscular blocking property (max with neomycin) as a side effect.
- Streptomycin
- Gentamycin
- Tobramycin
- Netilmicin
- Neomycin*
- Capreomycin*
- Kanamycin
- Amikacin
- Framycetin (soframycin)
* Capreomycin is chemically not an aminoglycoside, but have similar mechanism.
* Neomycin is used orally as gut sterilizer, to kill urease positive bacteria triggering hepatic coma.
* Streptomycin is used as 1st line anti-tubercular agent, while capreomycin, kanamycin and amikacin as 2nd line anti-tubercular agent.
* Framycetin is highly nephrotoxic like neomycin, hence not used for systemic administration. It is widely used topically for skin, eye and ear infections.
DNA gyrase inhibitors
DNA gyrase cuts double stranded DNA, introduces negative supercoils and then reseals the nicked ends. This is necessary step to prevent excessive positive supercoiling of the strands during transcription (making RNA copy of DNA sequence). Quinolones (nalidixic acid and fluroquinolones) act by inhibiting bacterial DNA gyrase. The bactericidal action probably results from digestion of DNA by exonucleases whose production is signaled by the damaged DNA.
Quinolones
quinolones are synthetic antimicrobials with quinolone structures that are particularly effective against gram-negative bacteria. Although the more recent fluorinated agents also inhibit gram-positive bacteria. The first member Nalidixic acid introduced in mid-1960s had usefulness limited to urinary and g.i. tract infections because of low potency, restricted spectrum and high frequency of bacterial resistance.
Fluorinated derivatives of quinolone developed in 1980s known as fluoroquinolones, which have high potency, a wider spectrum (gram-negative including pseudomonas and gram-positive including MRSA), a slower rate of resistance development, enhanced tissue penetration, and good tolerability. They are orally effective bactericidal agents. Fluroquinolones are contraindicated in pregnancy and child below 18 years (causes cartilage and tendon damage) and epileptic patients (it can induce seizure).
- Ciprofloxacin
- Ofloxacin
- Norfloxacin
- Levofloxacin
- Pefloxacin
- Sparfloxacin
- Moxifloxacin
- Gatifloxacin
- Trovafloxacin
* Ofloxacin, moxifloxacin, gatifloxacin, levofloxacin, are known as respiratory fluoroquinolones, because it kills wide range of bacteria causing respiratory tract infection.
metabolic inhibitors
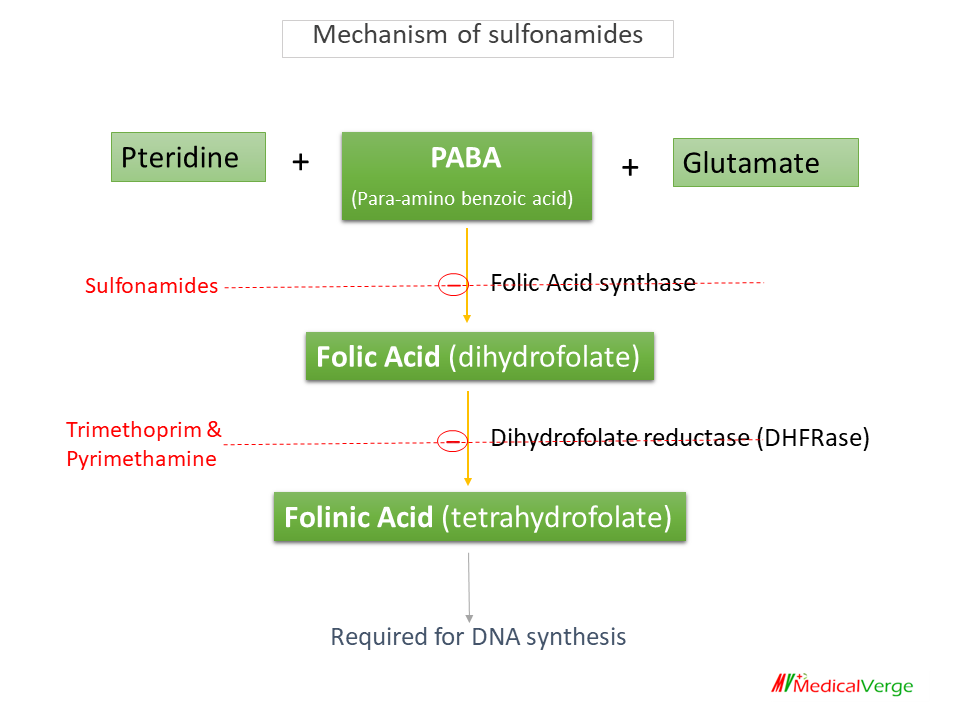
Sulfonamides
The first successful AMAs used to treat bacterial infections in humans were sulfonamides. They are primarily bacteriostatic against many gram-positive and gram-negative bacteria. Sulfonamides are structural analogue of PABA, competitively incorporated in place of PABA to inhibit Folic acid synthase enzyme and it prevents the formation of folic acid.
Sulfonamides have no effect on mammalian cells since they use folic acid from their diet rather than producing it. They are ineffective in the presence of pus as it is rich in PABA as well as purines and thymidine. Major adverse effects of sulfonamides include crystalluria, rash/ sulfa drug allergy, and SLE. it can cause kernicterus in newborn and hemolysis in G6PD-deficiancy patients.
- Sulfadoxine
- Sulfacytine
- Sulfamethoxazole
- Sulfisoxazole
- Sulfadiazine
- Sulfasalazine
- Dapsone
* Sulfadiazine is used as silver sulfadiazine for burn dressing.
* Sulfasalazine is a prodrug used for the treatment of ulcerative colitis as well as rheumatoid arthritis.
* Dapsone is used for the treatment of leprosy and dermatitis herpetiformis.
* Sulfadoxine is the longest acting while sufacytine is the shortest acting sulfonamide.
Dihydrofolate reductase (DHFRase) inhibitors
- Trimethoprim
- Pyrimethamine
They are not used alone, always used in combination with sulfonamides. Both, sulfonamide and DHFRase inhibitors are bacteriostatic alone, while combination is bactericidal. There are two FDCs (fixed dose combination) of DHFRase inhibitors –
* Cotrimoxazole (trimethoprim + sulfamethoxazole in 1:5 :: 80:400 mg), active against nocardia, burkholderia cepacia, and pneumocystis jirovacii.
* Sulfadoxine + Pyrimethamine, used in some parasitic diseases like malaria and toxoplasmosis.
Cell membrane inhibitors
Daptomycin
Daptomycin is a cyclic lipopeptide antibiotic produced from the bacterium Streptomyces roseosporus. Daptomycin binds to susceptible organisms’ cell membranes and causes a rapid depolarization of membrane potential. Intracellular DNA, RNA, and protein synthesis are inhibited as a result of membrane potential loss. It is bactericidal and administered by the intravenous route. Daptomycin is only active against gram-positive bacteria and now drug of choice for VRSA infections. Pulmonary surfactant inactivates daptomycin, therefore ineffective for the treatment of pneumonia; linezolid is preferred. Adverse effects include myopathy/rhabdomyolysis, eosinophilic pneumonia, and skin rash.
Polymyxin B
Polymyxin were obtained in the late 1940s from Bacillus polymyxa. It is a rapidly acting bactericidal agents, have a detergent-like action on the cell membrane. They have high affinity for phospholipids, causing membrane distortion or pseudopore formation.
Polymyxin E (colistin)
Colistin were obtained from B. colistinus. It has similar range of activity and mechanism as polymyxin B, but colistin is more potent on Pseudomonas, Salmonella and Shigella. It is bactericidal and effective against metallo-beta-lactamase producing bacteria.
* Polymyxin B and colistin, both are active against gram-negative bacteria only; all except Proteus, Serratia and Neisseria. They exhibit synergism with many other AMAs by improving their penetration into the bacterial cell. Resistance to these antibiotics has never been a problem. There is no cross resistance with any other AMA.